Bad Cancer ISGs in Immunotherapy
Chronic Interferon Signaling in Cancer Cells Promotes Immunotherapy Resistance
As described here, persistent interferon (IFN) signaling in cancer cells that occurs in the case of residual disease after immunotherapy can promote resistance or relapse. Here, IFN activates its receptor on cancer cells, enabling cancer cells to orchestrate immune dysfunction. Accordingly, deleting or blocking IFN signaling can restore response to immunotherapies such as immune checkpoint blockade (ICB).
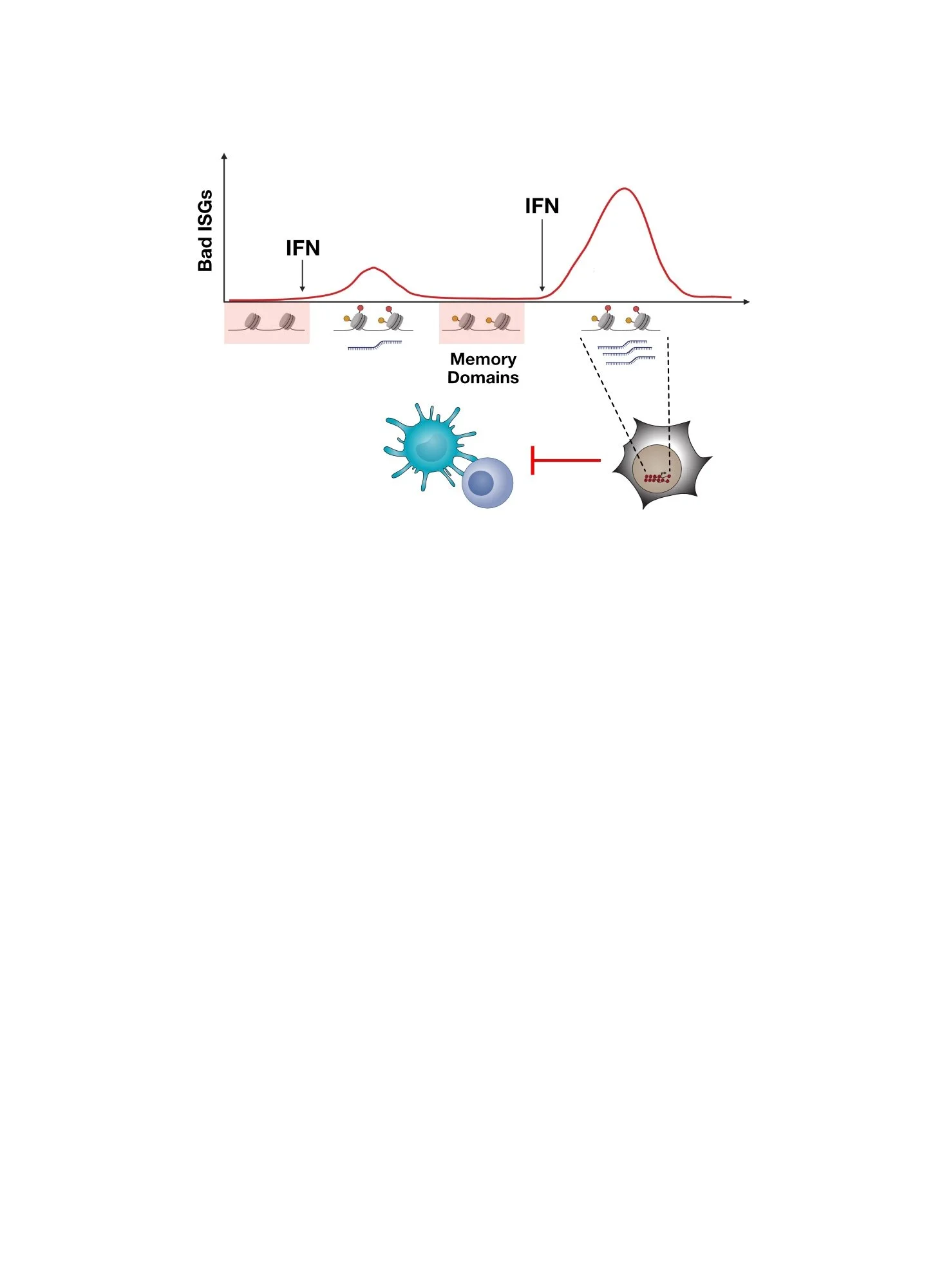
However, how does persistent IFN signaling in cancer cells orchestrate immune dysfunction? Typically IFN signaling activates hundreds of IFN-stimulated genes (ISGs). Which ISGs are “bad”, meaning which ones regulate resistance and immune dysfunction?
Chronic IFN signaling in cancer cells leads to high expression of cancer-associated ISGs and immune dysfunction. But which ISGs expressed by cancer cells are responsible for this feedback inhibition and how does this work?
RIPK1 is a “BAD” ISG Expressed by Cancer Cells
To investigate ISGs that might contribute to IFN-driven resistance, we examined which previously reported ICB resistance genes (Manguso et al., Nature 2017) are under the control of cancer cell IFN signaling. This revealed that many of these resistance genes are under the control of IFN signaling. However, one of the top IFNG-regulated ISGs in resistant cancer cells is RIPK1, implicating it in immunotherapy resistance. Moreover, human cancers have DNA copy number changes that increase RIPK1 gene expression, which is associated with worse patient survival. These observations suggest RIPK1 is a “bad” ISG.
What does RIPK1 do? RIPK1 is a kinase that regulates inflammation and how cells die upon stimulation of TNF superfamily (TNFSF) receptors. The inflammatory signaling arm is called complex I and is driven by the NFkB pathway. NFkB enhances cell survival and the production of multiple inflammation-related cytokines. In contrast, the cell death arm is called complex II and is comprised of proteins that promote cell death.
RIPK1 is an IFNG-regulated ISG in cancer cells. When IFNG signaling is blocked in ICB-resistant cancer cells, RIPK1 expression goes down. Across human tumors, RIPK1 levels increase due to DNA copy number aberrations. This is associated with worse patient survival.
In Resistant Cancer Cells, RIPK1 Directs TNFSF Signaling towards an NFkB Inflammatory and Cell Survival Response
In normal tissues, RIPK1 helps to ensure that inflammation after injury or infection is kept under control to prevent pathological conditions such as autoimmune or degenerative disease. But how do cancer cells take advantage of RIPK1? In contrast to normal tissues, cancer cells can use RIPK1 to direct TNFSF signaling predominantly through complex I and NFkB, resulting in overactive production of inflammatory cytokines and inappropriate cell survival. Thus, RIPK1 can confer several benefits for cancer cells including:
Increase in NFkB inflammatory cytokines such as CCL2 and CXCL1 that support the development of suppressive myeloid cells. This can create an immunosuppressive tumor microenvironment (TME).
Enhanced cell survival that makes cancer cells difficult to kill, particularly by immune cells. Although Perforin and Granzyme killing are widely appreciated, immune cells also use TNFSF signaling to kill cancer cells (e.g., TNF, TRAIL, FAS pathways). By diverting TNFSF signaling through complex I, RIPK1 actively enhances NFkB-mediated cell survival while circumventing complex II-mediated cell death.
RIPK1 directs TNFSF receptor signaling through complex I, which promotes NFkB-driven cell survival and suppressive cytokine production (follow red arrows in the pathway depiction, grey arrows indicate less active signaling). In the absence of RIPK1, NFkB activity decreases as complex II is favored. This inhibit suppressive cytokine production and sensitizes to cell death.
Deletion of RIPK1 in Cancer Cells Blocks Cell Extrinsic and Intrinsic Resistance to Immunotherapy
Given how cancer cells co-opt RIPK1 to enforce complex I and NFkB-driven cytokine production and survival signals, blocking RIPK1 could improve immunotherapy response. Indeed, across multiple tumor models, crippling RIPK1 can dramatically improve ICB efficacy. Tumors with RIPK1 deleted exhibit a marked decrease in distinct subsets of immunosuppressive myeloid cells, and are rendered sensitive to immune cell killing by the TNFSF pathway. Thus, RIPK1 is an ISG that controls a cancer cell extrinsic resistance mechanism — the accumulation of suppressive myeloid cells — and a cancer cell intrinsic resistance mechanisms — insensitivity to immune-mediated killing.
Finally, although RIPK1 is best known for its kinase function, we show that the ability of RIPK1 to control ICB resistance depends more on its scaffolding domain, which has implications for therapeutic strategies.
Multiple cytokines that promote the accumulation of ARG1+ suppressive myeloid cells are decreased in the absence of cancer cell RIPK1. This is accompanied by increased sensitivity to immune-mediated killing and improved ICB response.
Interested in learning more?
Visit our publications page for the full references to our work.
Cucolo et al., Immunity, 2022.
Here Are Some of the People That Led The Studies and Made It Happen!
Lisa Cucolo, former grad student and post-doc
Qingzhou Chen, post-doc in Junwei Shi’s lab